
CTX
Die cerebrotendinöse Xanthomatose, kurz CTX, ist eine erbliche Störung des Gallensäurestoffwechsels, die aufgrund von Genmutationen (im CYP27A1-Gen) verursacht wird. Cholesterin kann dadurch nicht in Gallensäuren umgewandelt werden, so dass es vermehrt zu Ablagerungen von Fetten (Cholesterin und Cholestanol) im Gehirn und in anderen Geweben kommt.1
CTX ist eine seltene Krankheit. Wie bei vielen seltenen Krankheiten ist davon auszugehen, dass die Krankheit weltweit unterdiagnostiziert ist. Wird die Krankheit frühzeitig erkannt und therapiert, können die Betroffenen ein nahezu normales Leben führen.
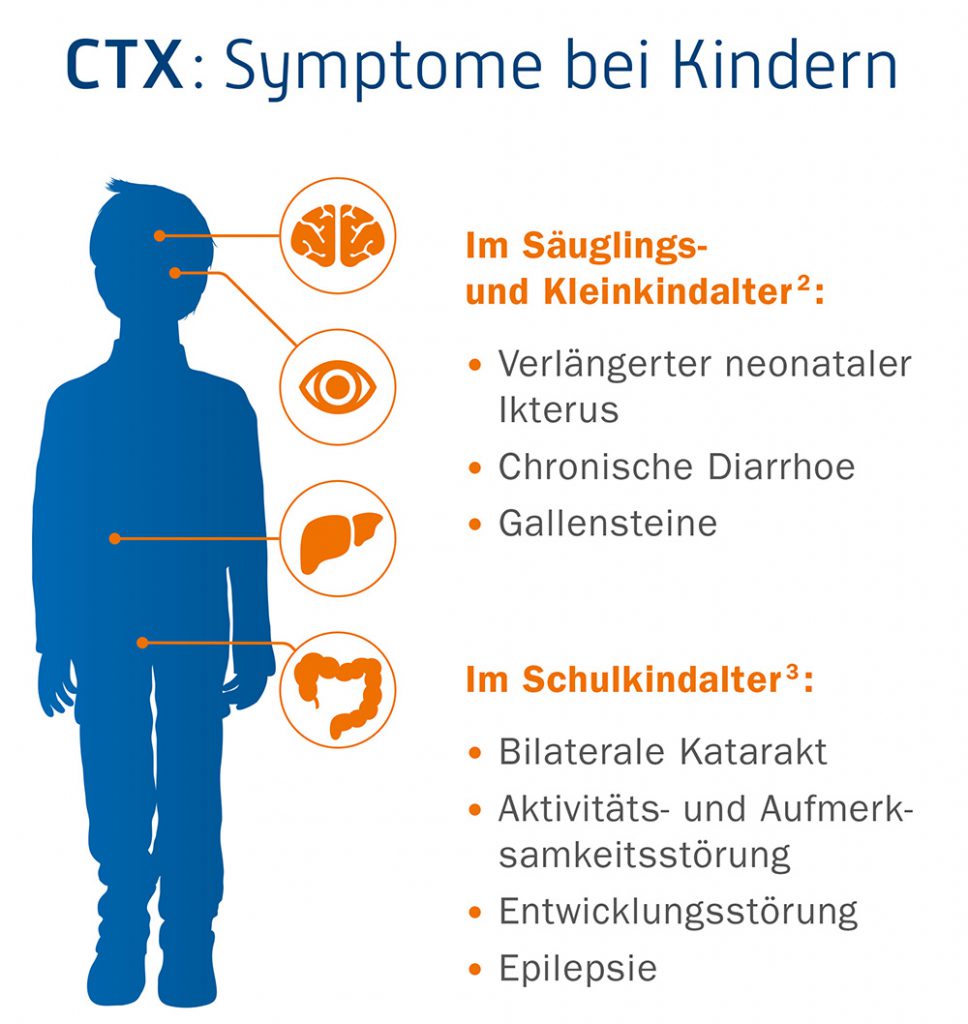
CTX kann sich gelegentlich schon bei Neugeborenen in einer verlängerten Neugeborenengelbsucht zeigen. Typische Symptome sind das Auftreten von chronischem Durchfall im Säuglingsalter in Verbindung mit einem – meist beidseitigem – Grauen Star (Linsentrübung) im Kindesalter. Aber auch Entwicklungsverzögerungen und neurologische Auffälligkeiten (Verhaltensauffälligkeiten, Lernschwäche) können ein Hinweis auf CTX sein. Unbehandelt treten bei den meisten Betroffenen im Jugendlichen- bzw. frühen Erwachsenenalter Xanthome (kleine gelblich-weiße Hautveränderungen) an der Achillessehne und an anderen Sehnen auf, und es entwickeln sich im Laufe der Zeit teilweise schwere neurologische und psychiatrische Störungen wie beispielsweise Demenz, Verhaltensauffälligkeiten, Dystonie, atypisches Parkinson-Syndrom, periphere Neuropathie und Krampfanfälle. Des Weiteren kann es zu frühzeitiger Arteriosklerose (Arterienverkalkung) und Osteoporose (Knochenschwund) kommen.
Eine frühe Diagnose ist für eine erfolgreiche Behandlung von CTX von entscheidender Bedeutung und kann für die Betroffenen – bei entsprechender Therapie – ein wichtiger Meilenstein hin zu einem nahezu normalen Leben ohne nennenswerte Symptome sein.
Ein wichtiger Schritt zur Diagnose von CTX ist die Bestimmung des Cholestanol-Plasmaspiegels (Fettwert im Blut). Ist dieser um das 3 bis 15-fache erhöht, ist dies bereits ein Hinweis auf CTX. Um die Diagnose abzusichern, ist der molekulargenetische Nachweis (Gendiagnostik) einer Mutation (Genveränderung) im CYP27A1-Gen empfehlenswert.4
Zur Behandlung der cerebrotendinösen Xanthomatose wird langfristig eine künstliche Gallensäure verabreicht, um die Konzentration von Cholestanol zu normalisieren und die Symptome sowie andere Begleiterkrankungen zu lindern. Manchmal werden zusätzlich Statine (Medikamente gegen erhöhte Blutfette) eingesetzt, um den Cholestanolwert zu senken und weitere Krankheitssymptome zu bekämpfen. Begleiterkrankungen wie Epilepsie, Spastizität und Parkinson werden symptomatisch behandelt. Oft ist eine Kataraktoperation erforderlich.5
M. Hodgkin
Morbus Hodgkin (auch Hodgkin-Lymphom oder Lymphogranulomatose genannt) zeigt sich durch einen bösartigen Tumor des Lymphsystems. Im Gewebe sind dabei sog. Sternberg-Reed-Zellen vorhanden. Diese entstehen aus den B-Lymphozyten (weiße Blutzellen), die ihre Keimzentren in den Lymphknoten haben.6 Die Sternberg-Reed-Zellen sind die bösartig wachsenden Zellen bei M. Hodgkin. Da sie jedoch nur etwa ein Prozent des Lymphoms ausmachen und der Rest durch reaktive Zellbeteiligung von CD4-positiven Lymphozyten, Monozyten, eosinophilen Granulozyten sowie Fibroblasten gebildet wird, ergibt sich bei M. Hodgkin ein „buntes“ zytologisches Bild.7
Die WHO unterscheidet in ihrer Klassifikation vier histologische Typen des so genannten klassischen Hodgkin-Lymphoms von einer weiteren Form, dem lymphozytenprädominanten Lymphom. Die vier unterschiedlichen Typen sind im Einzelnen:
- nodulär-sklerosierende Form (60 bis 80 Prozent der Fälle)
- gemischtzellige Form (15 Prozent der Fälle)
- lymphozytenreiche Form (drei bis vier Prozent der Fälle)
- lymphozytenarme Form (ein bis zwei Prozent der Fälle)
Die Ursache für die Krankheitsentstehung ist noch nicht ausreichend erforscht. Es wird aber vermutet, dass das sog. Epstein-Barr-Virus an der Krankheitsentstehung beteiligt sein könnte. Denn nach einem vorausgegangenen Pfeiffer’schen Drüsenfieber ist das Risiko, an M. Hodgkin zu erkranken, erhöht. Die Häufigkeit der jährlichen Neuerkrankungen (Inzidenz) des Hodgkin-Lymphoms beträgt zwei bis vier Erkrankungen pro 100.000 Personen, das Verhältnis von Männern zu Frauen liegt bei 3:2.8
Die Erkrankung zeigt sich zunächst durch schmerzlose Schwellungen von Lymphknoten. Sie treten v.a.am Hals, unter der Achsel oder in der Leistenregion auf. Zusätzlich kommt es zu einer unspezifischen Symptomatik, der sog. B-Symptomatik. Darunter fällt Fieber, Nachtschweiß, Leistungsminderung, Juckreiz und eine Gewichtsabnahme von mehr als zehn Prozent innerhalb von sechs Monaten. Im fortgeschrittenen Stadium kann es zu Störungen des Nervensystems, des Hormonhaushalts, des Urogenitaltrakts sowie zu Beschwerden bei Skelett- und Lungenbefall kommen. Eine Abschwächung des Immunsystems und infolgedessen häufige Infektionen sind möglich.
Diagnostiziert wird die Erkrankung durch eine Blutuntersuchung. Hierbei sind die Blutsenkungsgeschwindigkeit und das C-reaktive Protein (CRP) erhöht. Aber auch eine absolute Lymphopenie (Mangel an Lymphozyten), eine Eosinophilie, eine Anämie, eine Thrombopenie (Mangel an Blutplättchen) sowie eine LDH-Erhöhung. Diagnosesichernd sind Biopsien oder vollständig entnommene verdächtige Lymphknoten.
M. Hodgkin wird durch eine Kombination aus Chemotherapie und Bestrahlung therapiert. Dabei sind die Heilungsaussichten v.a. bei Kindern gut bis sehr gut. Die Chemotherapie wird dabei als Poly- oder Kombinationschemotherapie durchgeführt.
Seltener Hirntumor: Oligodendrogliom
Oligodendrogliome gehören zu der Gruppe der Gliome und entstehen aus Zellen, die im Gehirn das Stützgerüst – auch Glia genannt – bilden. Es handelt sich dabei um seltene Hirntumore, die gehäuft im Alter zwischen 35 und 50 Jahren auftreten.9
Es gibt zwei Formen der Erkrankung:
- gut differenziertes Oligodendrogliom, das langsam wächst (Klasse I)
- anaplastisches, schneller wachsendes Oligodendrogliom (Klasse II)
Die Ursache für die Entstehung eines Oligodendroglioms ist weitestgehend unbekannt. Normalerweise wachsen die Zellen im Zentralnervensystem (ZNS) kontrolliert und geordnet. Wird dieses kontrollierte Wachstum aus irgendeinem Grund gestört, beginnen diese Zellen sich zu teilen und einen Tumor zu bilden. Wie bei allen Tumorarten gibt es auch bei Oligodendrogliomen gutartige und bösartige Formen. Während gutartige Tumore weiter wachsen können ohne Beeinträchtigung des normalen Zellwachstums und mit milder Symptomatik, können bösartige Oligodendrogliome in Zellen eindringen, Gewebe zerstören und sich in andere Bereiche des Gehirns ausbreiten.9
Typische Symptome können zu Beginn der Erkrankung epileptische Anfälle sein. Aufgrund eines zu hohen Schädelinnendrucks können zudem Kopfschmerzen, Übelkeit (Erbrechen) und Sehstörungen hinzukommen. Ist die Krankheit bereits weiter fortgeschritten, kann der Patient durch die Symptome Bewusstseinsstörungen, Gangstörungen und Gedächtnisstörungen weiter beeinträchtigt werden.
Wichtig ist es, bei der Diagnosestellung möglichst zahlreiche Informationen über das Oligodendrogliom zu erhalten. Neben einfachen Reflextests, einer Untersuchung der Augenrückseite (Anophthamoscopie) gehört eine Computertomographie oder Magnetresonanztomographie zur ersten Diagnosestellung dazu. Diagnosesichernd ist jedoch nur eine histopathologische Untersuchung (Biopsie).
Die Behandlung eines Oligodendroglioms erfolgt in der Regel durch ein multidisziplinär aufgestelltes Spezialistenteam. Bevor ein Oligodendrogliom operativ entfernt wird, werden in der Regel Medikamente zur Senkung des Schädelinnendrucks verabreicht. Wegen seiner diffus infiltrierenden Natur kann ein Oligodendrogliom nicht vollständig entfernt werden und ist allein durch die Operation nicht heilbar. Daher kommen weitere Behandlungsmethoden wie eine Strahlentherapie, bei welcher hochenergetische Strahlen die Zerstörung der Krebszellen bewirken sollen, zum Einsatz. Außerdem kann ein Oligodendrogliom auch mittels Chemotherapie behandelt werden.
1 Pilo de la Fuente B, Ruiz I, Lopez de Munain A, Jimenez-Escrig A Cerebrotendinous xanthomatosis: Neuropathological findings. J. Neurol. 2008 May; 255 (6): 839–42. doi:10.1007/s00415-008-0729-6. PMID 18458861.
2 Steiner RD et al. http://emedicine.medscape.com/article/1418820-overview (aufgerufen am: 26.06.2020).
3 Frederico A und Dotti MT. Cerebrotendinous Xanthomatosis: Clinical Manifestations, Diagnostic Criteria, Pathogenesis, and Therapy, J of Child Neurology; Vol.18, 9, Sep 2003.
4 Nie S, Chen G, Cao X, Zhang Y. Cerebrotendinous xanthomatosis: a comprehensive review of pathogenesis, clinical manifestations, diagnosis, and management. Orphanet J Rare Dis. 2014 Nov 26;9:179. doi: 10.1186/s1023-014-0179-4. Review.PMID: 25424010
5 Medicine, U. S. N. L. o. Cerebrotendinous xanthomatosis, http://ghr.nlm.nih.gov/condition/cerebrotendinous-xanthomatosis (201) [Letzter Zugriff am 11.03.2020].
6 J. S. Knight u. a.: Epstein-Barr virus latent antigen 3C can mediate the degradation of the retinoblastoma protein through an SCF cellular ubiquitin ligase. In: PNAS. (2005) 102(51), S. 18562–18566, PMID 16352731, PMC 1317900 (freier Volltext)
7 Henry H. Balfour Jr.: Progress, prospects, and problems in Epstein-Barr virus vaccine development. In: Current Opinion in Virology. Band 6, Juni 2014, ISSN 1879-6257, S. 1–5, doi:10.1016/j.coviro.2014.02.005.
8 https://de.wikipedia.org/wiki/Hodgkin-Lymphom (Zugriff am 11.03.2020)
9 https://www.hirntumorhilfe.de/hirntumor/tumorarten/oligodendrogliom/ (Zugriff am 11.03.2020)